
14 8.10 Alkynes
Chapter 8.10 Learning Objectives
- Be able to prepare alkynes.
- Be able to extend the alkyne using chain lengthening reactions.
- Be able to predict the products of the following reaction with alkynes:
- Hydrohalogenation
- Hydration
- Halogenation
- Reduction
- Oxidation
- Oxidative cleavage
In this section, we will explore the methods for the synthesis of alkyne and the chemical reactions of alkynes.
8.10.1 Acidity of Terminal Alkynes and Related Reactions
In the discussions of acids and bases (Chapter 3), we have learned that the hydrogen atom bonded to the terminal alkyne carbon shows higher acidity than the hydrogen atoms bonded to the carbons of an alkene or alkane, and the pKa value of the terminal alkyne hydrogen is about 25.
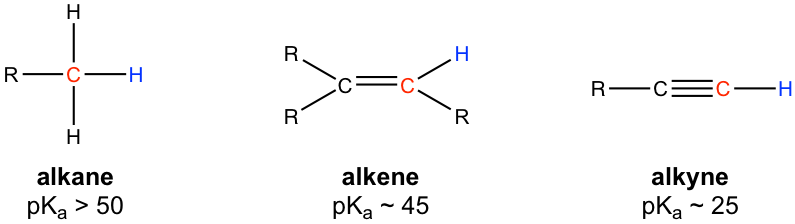
Because of the relative high acidity, the terminal alkynes can be deprotonated by appropriate strong bases, such as NaH, NaNH2.
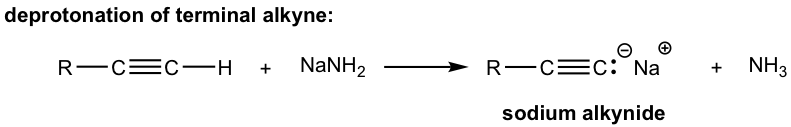
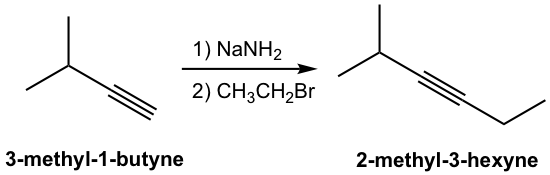
The product of the above deprotonation, alkynide anion, is a good nucleophile that can be used in SN2 reaction with primary substrates (since primary substrates work best for such SN2 reaction as we have learned):

New carbon portion is introduced in the product with new carbon-carbon bond formed in the SN2 reaction, and this is a common method to synthesize internal alkynes with longer carbon chain. A specific example for the synthesis of 2-methyl-3-hexyne from 3-methyl-1-butyne is given here:
Key Takeaways
Terminal alkynes can form acetylide anions with a strong base such as sodium hydride. The acetylide anion can be reacted with primary alkyl halides via a SN2 reaction leading to lengthening of the alkyne.
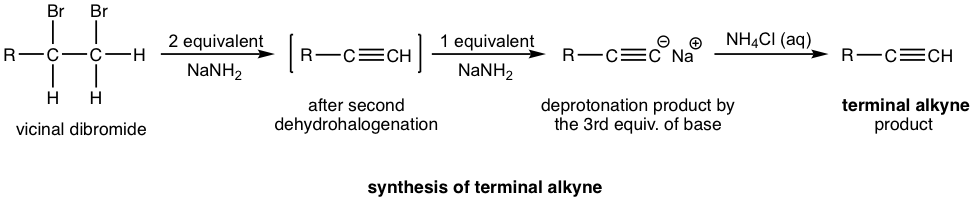
The more general way to synthesize alkyne is via the elimination reaction of vicinal dihalides. Recall that vicinal dihalides are the halogenation products of alkenes (section 10.4). The vicinal dihalide can then be subjected to a double dehydrohalogenation reaction with a strong base to produce an alkyne.
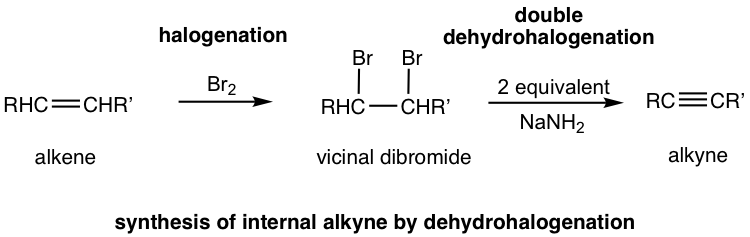
The dehydrohalogenation occurs twice, in two steps, the first product is a haloalkene, and the second product is the alkyne. Amide, usually NaNH2, is a base that is strong enough to cause both reactions carried out consecutively in the same mixture. Two molar equivalents of sodium amide per mole of the dihalide are required to ensure the elimination occur two times.

If a terminal alkyne is the desired product, then three molar equivalents of base are required. The terminal alkyne produced after double dehydrohalogenation is deprotonated by sodium amide, the third mole of base is to ensure the deprotonation occurs completely and all the terminal alkyne converted to the salt format. The salt of alkynide was then treated with ammonium chloride (or water, as source of proton) to produce terminal alkyne as the final desired product.
Here is another example:
Alkynes can be prepared by the elimination of HX from alkyl halides in a similar manner as alkenes. Treatment of a 1,2-dihaloalkane (called a vicinal dihalide) with an excess amount of a strong base such as KOH or NaNH2 results in a twofold elimination of HX and formation of an alkyne. The starting vicinal dihalides are themselves readily available by addition of Br2 or Cl2 to alkenes. Thus, the overall halogenation/dehydrohalogenation sequence makes it possible to go from an alkene to an alkyne. For example, diphenylethylene is converted into diphenylacetylene by reaction with Br2 and subsequent base treatment. Thus, the overall halogenation/dehydrohalogenation sequence makes it possible to go from an alkene to an alkyne. For example, diphenylethylene is converted into diphenylacetylene by reaction with Br2 and subsequent base treatment.

Examples
Design the synthesis route of 1-butyne from 1-butene.
Approach:
Use retro-synthetic analysis:
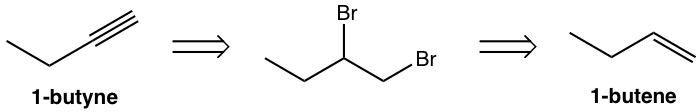
That analysis can be translated to the step-by-step synthesis as:
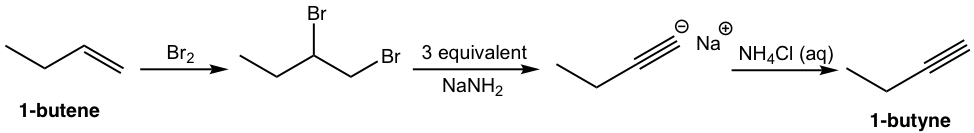
Solution: (show the steps together in the proper order, without showing intermediates for each step)
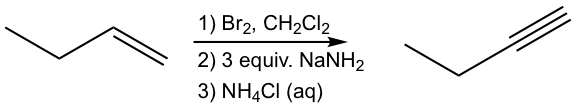
Key Takeaways
Alkenes reacts with bromine to form dibromides. The dibromides can undergo double elimination to form alkynes.
8.10.3 Reactions of Alkynes
8.10.3.1 Hydrogenation of Alkynes
The catalytic hydrogenation applied to the π bonds of C≡C triple bonds as well. Depending on the conditions and catalysts employed, one or two molar equivalents of hydrogen will be added to a triple bond and alkene or alkane produced as the product respectively.
When platinum or palladium catalysts applied, the final product of the hydrogenation is an alkane with sufficient hydrogen provided. The initial product is an alkene, that undergoes the reaction successively to give alkane as the final product.
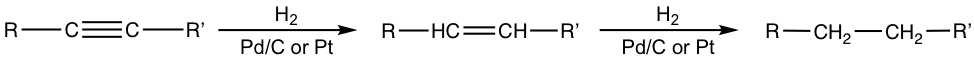
With certain catalyst used, the hydrogenation of alkyne can be stopped at the alkene stage. The most commonly employed catalyst is the Lindlar catalyst. Lindlar catalyst is prepared by precipitating palladium on calcium carbonate and then treating it with lead (II) acetate and quinoline. The special treatment modifies the surface of the palladium metal by partially deactivating it, and making it more effective at catalyzing the hydrogenation to a triple bond rather than to a double bond.
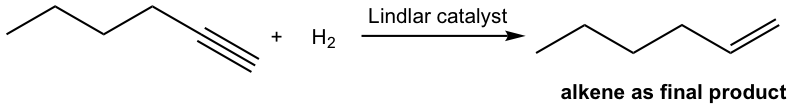
The mechanism for the catalytic hydrogenation of alkyne is almost the same as that of alkene. Since both hydrogen atoms are delivered from the surface of the catalyst, they are delivered to the same side of the triple bond, therefore the syn addition occurs. So the hydrogenation of an internal alkyne produces cis-alkene with the Lindlar catalyst.
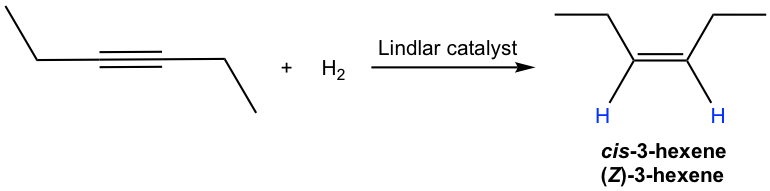
Internal alkyne can be converted into trans-alkene using sodium (or lithium) in liquid ammonia. The mechanism for this reaction involves successive single electron transfers from the metal (sodium or lithium) and proton transfers from ammonia, with radical intermediates. The sodium metal (or lithium) reacts more rapidly with triple bond than double bond, so the reaction stops at the alkene stage. Low temperature (-78 °C) is necessary to keep ammonia at the liquid state.
The trans-vinylic anion is formed preferentially because of the higher stability with two R groups farther apart. Protonation of the trans-vinylic anion leads to the trans-alkene.

The alkyne hydrogenation reaction has been explored extensively by the Hoffmann–LaRoche pharmaceutical company, where it is used in the commercial synthesis of vitamin A. The cis isomer of vitamin A produced initially on hydrogenation is converted to the trans isomer by heating.

Key Takeaways
- Alkynes can be reduced to alkanes using hydrogen and a metal catalyst.
- Alkynes can be reduced to cis-alkenes by Lindlar catalyst.
- Alkynes can be reduced to trans-alkenes by sodium (or potassium or lithium) and liquid ammonia.
8.10.3.2 Hydrohalogenation of Alkynes
An alkyne is an electron-rich molecule with high density of pi electrons, therefore it is a good nucleophile that reacts readily with electrophiles. Thus alkynes, like alkenes, also undergo electrophilic addition with hydrogen halide.
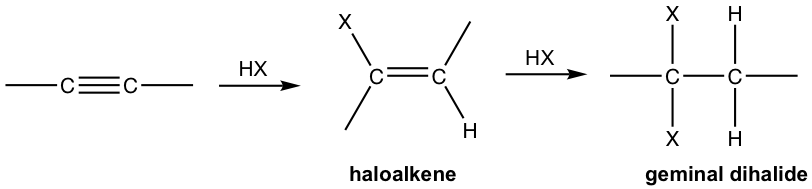
- Alkyne reacts with one mole of HX to form haloalkene, and with two moles of HX to form geminal dihalides, the dihalide with both halogen attached to the same carbon. “Geminal” comes from geminusin Latin, that means “twin”.
- Both addition follow Markovnikov’s rule in terms of regioselectivity.
If one molar equivalent of HX available, the addition can be stopped at the first addition to haloalkene. The halo-substituted alkene is less reactive than alkyne for electrophilic addition because a halogen substituent withdraws electrons inductively, therefore decreasing the nucleophilicity of the double bond.
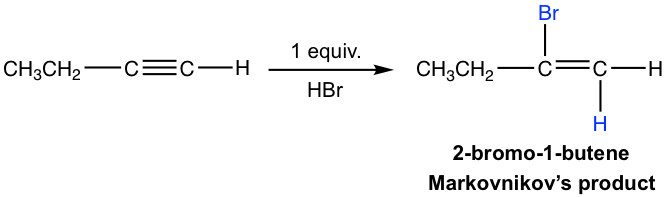
The mechanism for the electrophilic addition to alkyne is rather similar to the addition of alkene, with protonation as the first step. For terminal alkyne, if the protonation occurs on different triple bond carbon, the primary or secondary vinylic cation intermediate will formed. The higher stability of the secondary vinylic cation leading to the Markovnikov’s regioselectivity, that the hydrogen atom attached to the carbon that has the greater number of hydrogen atoms.

If excess hydro halide is present, the addition to alkyne occur twice to give geminal halide that follow the Markovnikov’s regioselectivity.
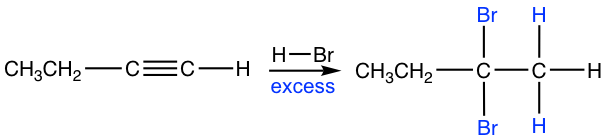
Key Takeaways
Alkynes react with 2 moles of HX to form geminal dihalides. The addition is regioselective and follows Markovnikov’s rule.
8.10.3.3 Hydration of Alkynes
Alkynes also undergo the acid-catalyzed addition of water (hydration), similar to alkenes. As a result, the H added to one triple bond carbon and OH added to the other triple bond carbon, and the product formed is called an enol (“en” comes from “ene” that means double bond, “ol” means OH group). An enol is a compound with a carbon-carbon double bond and an OH group connected on one of the double bond carbon.
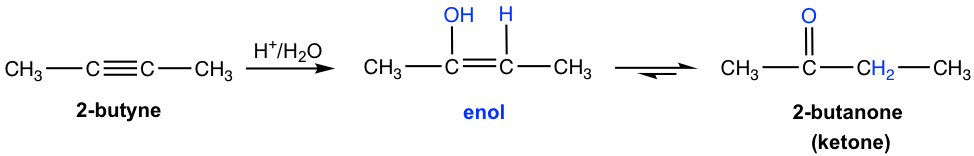
Enol is a very unstable compound, it immediately undergoes rearrangement to give more stable carbonyl compound, aldehyde or ketone. The structure of a carbonyl compound and an enol differ in the location of the double bond and a hydrogen atom, and they are called tautomers. The interconversion between the tautomers is called tautomerization. The mechanism is not covered. Enol always undergoes tautomerization rapidly because of the high stability of carbonyl compound, as shown in the general way below.

For symmetrical internal alkyne that has the same group attached to each of the triple bond (sp) carbon, the addition of water forms a single ketone as a product. As in the early example that 2-butanone is produced from the hydration of 2-butyne.
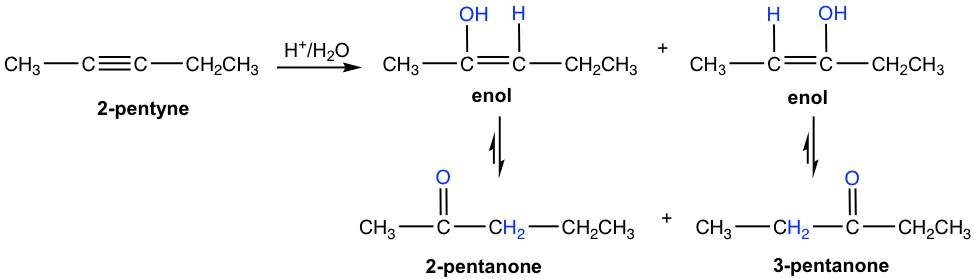
For unsymmetrical internal alkyne with different groups on each of the triple bond carbon, the mixture of two ketones are formed because the initial addition of the proton can occur on either of the sp carbons. The hydration of 2-pentyne is shown here that produce the mixture of 2-pemtanone and 3-pentanone as product.
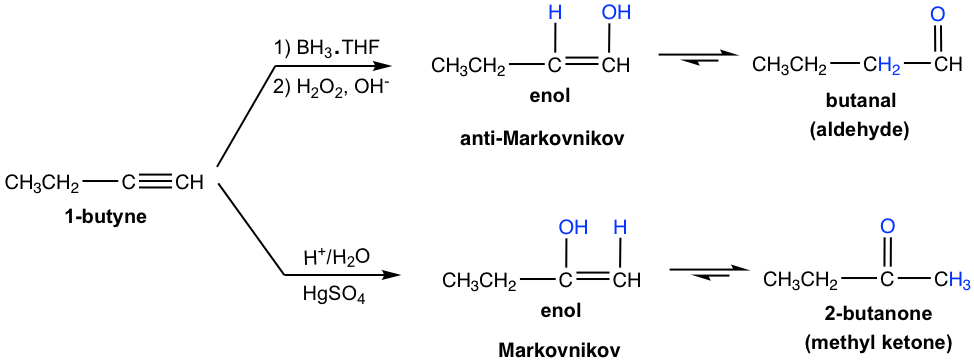
Terminal alkynes are not as reactive as internal alkynes towards the hydration. The addition of water to a terminal alkyne will occurs if mercuric ion (Hg2+) present as a catalyst. The enol formed from the addition follows the Markovnikov’s rule with the hydrogen atom attached to the terminal carbon, and a methyl ketone (the ketone with a methyl group connected on one side of the C=O bond) is the final product after tautomerization.
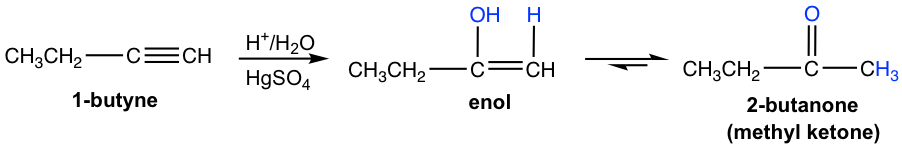
The mechanism of hydration of alkynes is shown here:

8.10.3.4 Hydroboration-Oxidation of Alkynes:
Hydroboration-oxidation also applies to alkyne in the similar way as to alkene. The two-step process results in the enol, that goes through tautomerization to give carbonyl compound.
Meanwhile, the addition of borane to a terminal alkyne shows the same regioselectivity as observed in borane addition to an alkene. That is boron adds preferentially to the terminal triple bond (sp) carbon (the carbon with more hydrogen atom), or the terminal carbon with less substituents. After oxidation, the boron-containing group is converted to the OH group, so the enol is produced in the anti-Markovnikov way, with OH connected on the terminal carbon. The tautomerization of such enol generates aldehyde as the final product.
Comparing the two hydration methods of alkyne, hydroboration-oxidation produces aldehyde from terminal alkyne, while acid-catalyzed hydration converts terminal alkyne to methyl ketone.
Key Takeaways
- Water adds to alkynes to form enols. Enols are unstable and undergo keto-enol tautomerization to form carbonyl compounds.
- Oxymercuration of both internal and terminal alkynes forms ketones.
- Hydroboration of internal alkynes forms ketones while that of terminal alkynes form aldehydes.
- Addition of water is regioselective. Oxymercuration follws Markovnikov’s rule while hydroboration results in anti-Markovniko’s orientation.
- Oxymercuration proceeds via mercurinium ion intermediate and the addition of water is anti.
- Hydroboration is concerted and the addition of water is syn.
Exercises: Synthesis and Reactions of Alkynes
- Synthesis of Alkynes Worksheet Synthesis of Alkynes Worksheet Key
- Predict the product for each of the following reactions:
- Predict the correct reagent that can bring about each transformation:
